Bioinformatics (BI)
1.Definition and value of bioinformatics
At the end of the 20th century, the completion of the Human Genome Project marked a milestone in scientific history providing a nearly complete blueprint of human genetic information.
Although this achievement—comprising approximately three billion base pairs—represented one of the greatest scientific accomplishments of humankind, it merely provided access to a vast repository of biological data encoding the blueprint of life. Without proper interpretation, the fundamental nature of life and the underlying causes of disease remain largely unresolved.
To address this challenge, the field of bioinformatics has emerged as a critical discipline that integrates computer science, statistics and biology to analyze large-scale biological data elucidate biological principles and uncover disease mechanisms.
With the rapid advancement of next-generation sequencing (NGS) technologies, the volume of genomic data has increased exponentially. Diverse types of biological data—including genomic variants, copy number aberrations, DNA methylation, histone modifications and single-cell transcriptomic data—are generated on a daily basis. The ability to analyze and interpret these complex datasets has become a fundamental competency in modern life sciences research.
Our research focuses on leveraging large-scale biological data to understand the molecular basis of diseases and to identify novel therapeutic strategies. In particular, bioinformatics plays a pivotal role in cancer research. We utilize genomic data to identify tumor-driving mutations, investigate tumor evolution and develop personalized therapeutic strategies within the framework of precision medicine.
Bioinformatics is not merely a tool for data analysis but rather a new scientific language that enables us to decode the complexity of biological systems and shape the future of medicine.
2. Bioinformatics research strategies in our lab
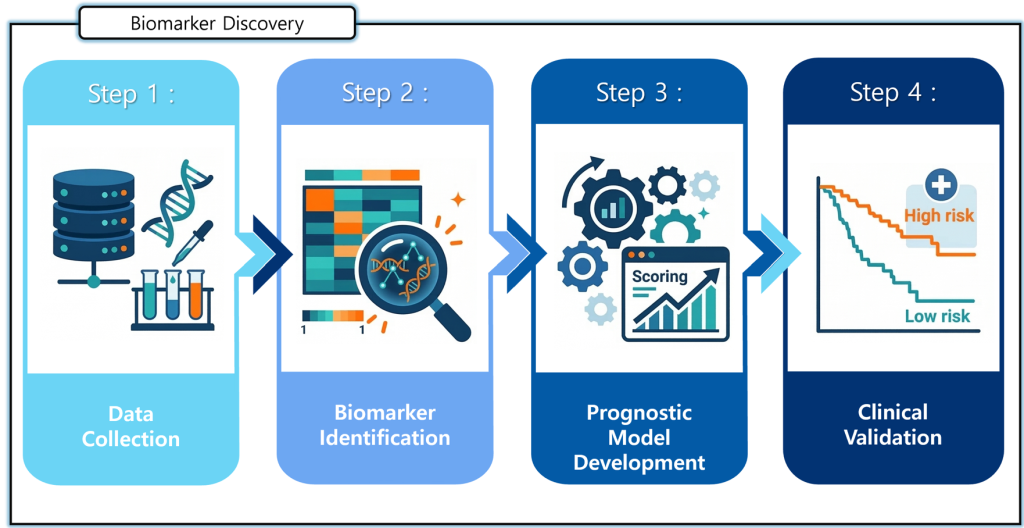
2-1) Biomarker discovery and validation for clinical outcome prediction
The schematic on the left illustrates the overall workflow of our research aimed at identifying biomarkers that can predict disease prognosis and validating their clinical applicability.
In the data collection stage, diverse biological data—including patient samples, genomic information and experimental results—are systematically collected. In the biomarker identification stage, these data are analyzed to identify key genes or molecular signatures associated with disease progression and patient outcomes.
Subsequently, in the prognostic model development stage, predictive models are constructed based on the identified biomarkers to estimate the likelihood of disease progression. Finally, in the clinical validation stage, the developed models are applied to independent patient cohorts to stratify patients into high- and low-risk groups and to evaluate their predictive performance.
Through this research framework, we aim to develop novel biomarkers and predictive models that enable more accurate prognosis and support optimized, patient-specific therapeutic strategies.d
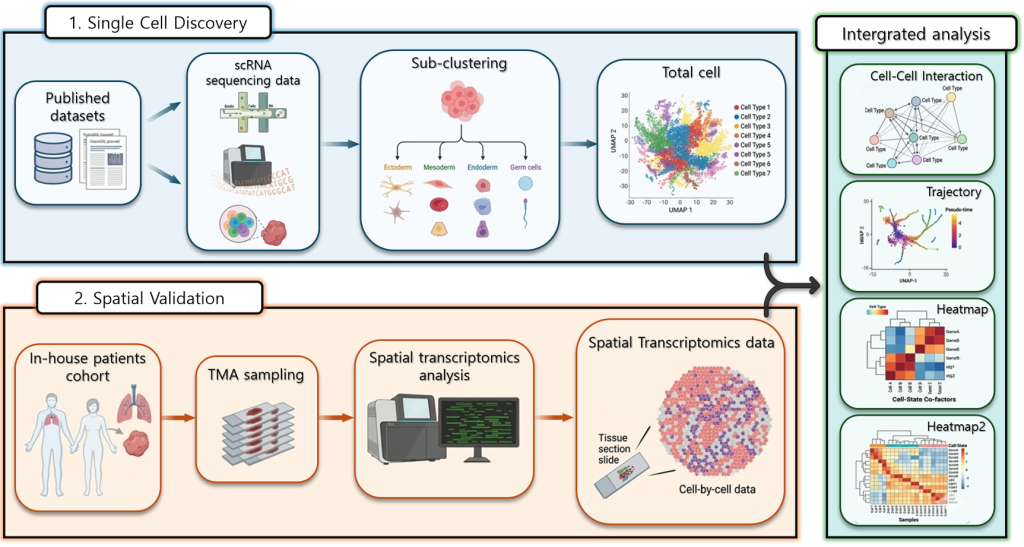
2-2) Integrated analysis of cellular composition and cell–cell interactions ssing single-cell and spatial transcriptomics
The schematic on the right illustrates the overall analytical workflow integrating single-cell and spatial transcriptomic analyses to investigate cellular characteristics and interactions within diseased tissues.
In the single-cell discovery stage, publicly available single-cell RNA sequencing data are analyzed to characterize gene expression profiles at the cellular level. This enables the identification and classification of diverse cell populations within the tissue as well as the characterization of distinct cell types and their molecular features.
In the spatial validation stage, patient-derived tissue samples are used to validate the spatial distribution of the identified cellular features. Tissue microarrays (TMAs) are prepared from patient specimens followed by spatial transcriptomic analysis to determine the localization of individual cells within the tissue. This approach allows for the integration of cell-type–specific gene expression profiles with their spatial context.
Finally in the integrated analysis stage, single-cell and spatial transcriptomic datasets are combined to perform comprehensive analyses including cell–cell interactions, cellular trajectories and gene expression dynamics.
Through this integrated framework, we aim to achieve a deeper understanding of how cellular interactions within the tissue microenvironment contribute to disease progression.